|
|

A
32-year-old woman presents to the emergency department with
several flesh-colored papules on her face, trunk, and upper
extremities. She noticed the lesions at approximately 10
years of age. However, over the past 5 years, the lesions
have increased in number and become uncomfortable. She
primarily complains of irritation from the lesions along
her bra line. She previously underwent excision of other
similar skin lesions 5 years ago, but these have since
recurred. She denies having discharge, pain, trauma,
contact with individuals with atypical skin lesions or
rashes, travel out of the country, unusual exposure to
animals, or a history of sexually transmitted disease.
The patient's medical and surgical history includes
environmental allergies, frequent episodes of bronchitis,
and the aforementioned excisions. She takes cetirizine HCl
(Zyrtec) and fluticasone propionate (Flonase) for allergies
and has no known drug allergies. Her family history is
significant for coronary artery disease, hypertension,
diabetes mellitus, and glaucoma. She does not smoke and
drinks alcohol on occasion. The review of systems is
otherwise noncontributory.
Physical examination reveals dozens of fleshy nodules of
0.5-2.0 cm throughout her trunk, face, and upper
extremities. The nodules are nontender to palpation and
nonerythematous, and they produce no discharge, crusting,
or scaling. Several 1.5- to 3-cm, tan, oval macules and
patches with well-defined borders are located on her trunk
and upper extremities (see Images). Her vital signs are
within normal limits, and the rest of the physical findings
are unremarkable.
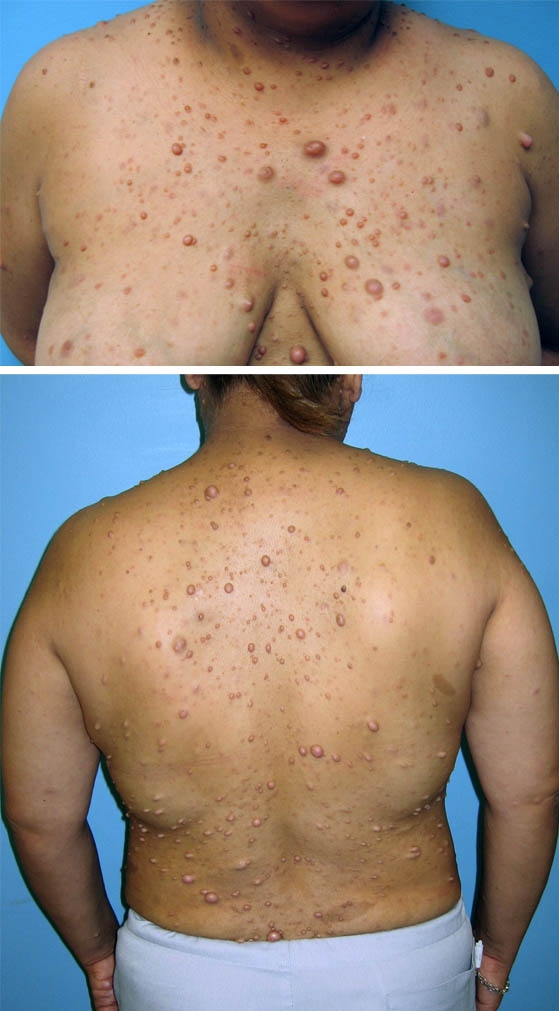
What is the diagnosis?
Answer
Neurofibromatosis:
Neurofibromatosis (NF) is an autosomal dominant disorder
with numerous presentations affecting nearly every organ
system. The 2 major subtypes are NF type 1 (NF1), also
known as peripheral NF, and NF type 2 (NF2), referred to as
central NF. However, these terms are not completely correct
because NF1 may cause central characteristics.
About 50% of cases of NF are familial, and the other 50%
are due to spontaneous gene mutation. NF1, also known as
von Recklinghausen disease, is a common genetic disorder
involving a gene mutation on chromosome 17 that affects 1
in every 3000-4000 births (Children's Tumor Foundation,
2006). This disorder affects all races and both sexes
equally (Neurofibromatosis, Inc, 2006). The diagnosis of
NF1 requires that the patient present with 2 or more of the
following conditions: 6 or more café au lait spots
(irregularly shaped, evenly pigmented, brown macules), 2 or
more neurofibromas, axillary or inguinal freckling, Lisch
nodules on the iris, optic glioma, various types of osseous
lesions, or a first-degree relative with the condition.
Symptomatic NF1 typically manifests as flesh-colored,
benign skin tumors that appear late in childhood. The
patient may have as few as 3 or as many as thousands of
these benign lesions, which consist of Schwann cells,
neural fibroblasts, mast cells, and vascular elements.
Neurofibromas may occur anywhere in the body and
potentially lead to marked disfigurement. Lesions along
visual, auditory, or CNS nerve pathways may result in
blindness, deafness, or neurologic deficits. Other findings
associated with this condition include skeletal anomalies,
such as fibrous dysplasia, subperiosteal bone cysts, or
vertebral scalloping (Neurofibromatosis, Inc, 2006).
NF2 is a progressive genetic disorder affecting 1 in every
33,000-40,000 births (Neurofibromatosis, Inc, 2006).
Patients with NF2, which results from an abnormality of
chromosome 22, typically present with acoustic neuromas or
vestibular schwannomas. Clinical manifestations include
tinnitus, balance disorders, and progressive hearing loss.
Affected patients may also have meningiomas and juvenile
cataracts. The diagnosis is based on a history of the
condition in a first-degree relative and on any 2 of the
conditions listed above for NF1 (Neurofibromatosis, Inc,
2006).
For both NF1 and NF2, the diagnosis is primarily based on
physical findings and a positive family history. Diagnostic
tests that may be useful include radiographic studies, such
as CT of the brain, genetic analysis, and psychological or
developmental assessment.
No cure exists for this condition. Recommendations for
follow-up include referral to support groups, psychological
counseling, evaluation of learning disorders, surgical
excision of lesions, and regular monitoring by a primary
care provider for any changes that may occur, as patients
with NF1 are at somewhat increased risk of malignancy.
Annual ocular examinations are recommended. Genetic testing
is also advocated in patients with NF who wish to have
children. Surgery has been a successful treatment for the
lesions themselves; however, recurrence often occurs, and
nerve damage is a risk when tumors are located along neural
pathways (National Institute of Neurologic Disorders and
Stroke, 2006).
References
|
Link
to further Information on:

For
more information on neurofibromatosis, see the eMedicine
articles Neurofibromatosis
(within the Dermatology specialty), Neurofibromatosis,
Type 1 and Neurofibromatosis,
Type 2 (within the Neurology specialty), and Neurofibromatosis
Type 1 and Neurofibromatosis
Type 2 (within the Radiology specialty). For other
resources and access to support networks, contact the Web
pages of Children's
Tumor Foundation and Neurofibromatosis,
Inc.
|
|



 DISCLAIMER:
This website is designed primarily for use by qualified
physicians and other medical professionals. The
information provided here is for educational and
informational purposes only. It is not guaranteed to be
correct and should NOT be considered as a substitute for
the advice of an appropriately qualified expert. In no way
should the information on this site be considered as
offering advice on patient care decisions or establishment
of a patient-physician relationship.
DISCLAIMER:
This website is designed primarily for use by qualified
physicians and other medical professionals. The
information provided here is for educational and
informational purposes only. It is not guaranteed to be
correct and should NOT be considered as a substitute for
the advice of an appropriately qualified expert. In no way
should the information on this site be considered as
offering advice on patient care decisions or establishment
of a patient-physician relationship.